| Index to this page |
In the living cell, DNA undergoes frequent chemical change, especially when it is being replicated (in S phase of the eukaryotic cell cycle). Most of these changes are quickly repaired. Those that are not result in a mutation. Thus, mutation is a failure of DNA repair.
| Link to discussion of DNA repair. |
A single base, say an A, becomes replaced by another. Single base substitutions are also called point mutations. (If one purine [A or G] or pyrimidine [C or T] is replaced by the other, the substitution is called a transition. If a purine is replaced by a pyrimidine or vice-versa, the substitution is called a transversion.)
With a missense mutation, the new nucleotide alters the codon so as to produce an altered amino acid in the protein product.
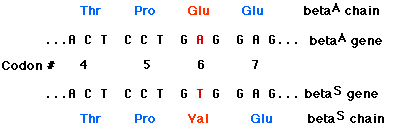
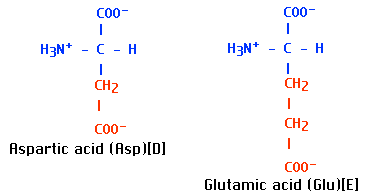
EXAMPLE: sickle-cell disease. The replacement of A by T at the 17th nucleotide of the gene for the beta chain of hemoglobin changes the codon GAG (for glutamic acid) to GTG (which encodes valine). Thus the 6th amino acid in the chain becomes valine instead of glutamic acid.
ANOTHER EXAMPLE: Patient A with cystic fibrosis (scroll down).
With a nonsense mutation, the new nucleotide changes a codon that specified an amino acid to one of the STOP codons (TAA, TAG, or TGA). Therefore, translation of the messenger RNA transcribed from this mutant gene will stop prematurely. The earlier in the gene that this occurs, the more truncated the protein product and the more likely that it will be unable to function.
EXAMPLE: Patient B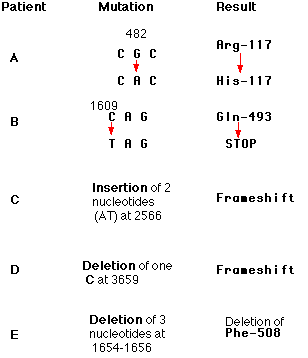
Here is a sampling of mutations that have been found in patients with cystic fibrosis. Each of these mutations occurs in a huge gene that encodes a protein (of 1480 amino acids) called the cystic fibrosis transmembrane conductance regulator (CFTR). The protein is responsible for transporting chloride and bicarbonate ions through the plasma membrane. The gene encompasses over 188,000 base pairs on chromosome 7 embedded in which are 27 exons encoding the protein. The numbers in the mutation column represent the number of the nucleotides affected. Defects in the protein cause the various symptoms of the disease [More]. Unlike sickle-cell disease, then, no single mutation is responsible for all cases of cystic fibrosis. People with cystic fibrosis inherit two mutant genes, but the mutations need not be the same.
In one patient with cystic fibrosis (Patient B), the substitution of a T for a C at nucleotide 1609 converted a glutamine codon (CAG) to a STOP codon (TAG). The protein produced by this patient had only the first 493 amino acids of the normal chain of 1480 and could not function.
Most amino acids are encoded by several different codons. For example, if the third base in the TCT codon for serine is changed to any one of the other three bases, serine will still be encoded. Such mutations are said to be silent because they cause no change in their product and cannot be detected without sequencing the gene (or its mRNA).
The removal of intron sequences, as pre-mRNA is being processed to form mRNA, must be done with great precision. Nucleotide signals at the splice sites guide the enzymatic machinery. If a mutation alters one of these signals, then the intron is not removed and remains as part of the final RNA molecule. The translation of its sequence alters the sequence of the protein product.
| Link to discussion of RNA processing. |
Extra base pairs may be added (insertions) or removed (deletions) from the DNA of a gene. The number can range from one to thousands. Collectively, these mutations are called indels.
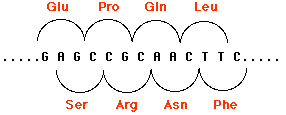
Indels involving one or two base pairs (or multiples of two) can have devastating consequences to the gene because translation of the gene is "frameshifted". This figure shows how by shifting the reading frame one nucleotide to the right, the same sequence of nucleotides encodes a different sequence of amino acids. The mRNA is translated in new groups of three nucleotides and the protein specified by these new codons will be worthless. Scroll up to see two other examples (Patients C and D).
Frameshifts often create new STOP codons and thus generate nonsense mutations. Perhaps that is just as well as the protein would probably be too garbled anyway to be useful to the cell.
Indels of three nucleotides or multiples of three may be less serious because they preserve the reading frame (see Patient E above). Over a thousand different mutations have been found in the CFTR gene, but Patient E's deletion is by far the most common.
However, a number of inherited human disorders are caused by the insertion of many copies of the same triplet of nucleotides. Huntington's disease and the fragile X syndrome are examples of such trinucleotide repeat diseases.
Several disorders in humans are caused by the inheritance of genes that have undergone insertions of a string of 3 or 4 nucleotides repeated over and over. A locus on the human X chromosome contains such a stretch of nucleotides in which the triplet CGG is repeated (CGGCGGCGGCGG, etc.). The number of CGGs may be as few as 5 or as many as 50 without causing a harmful phenotype (these repeated nucleotides are in a noncoding region of the gene). Even 100 repeats usually cause no harm. However, these longer repeats have a tendency to grow longer still from one generation to the next (to as many as 4000 repeats).
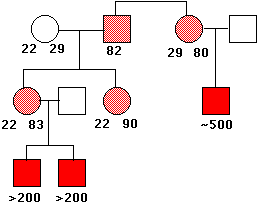
This causes a constriction in the X chromosome, which makes it quite fragile. Males who inherit such a chromosome (only from their mothers, of course) show a number of harmful phenotypic effects including mental retardation. Females who inherit a fragile X (also from their mothers; males with the syndrome seldom become fathers) are only mildly affected.
This image shows the pattern of inheritance of the fragile X syndrome in one family. The number of times that the trinucleotide CGG is repeated is given under the symbols. The gene is on the X chromosome, so women (circles) have two copies of it; men (squares) have only one. People with a gene containing 80–90 repeats are normal (light red), but this gene is unstable, and the number of repeats can increase into the hundreds in their offspring. Males who inherit such an enlarged gene suffer from the syndrome (solid red squares). (Data from C. T. Caskey, et al.).
Some forms of muscular dystrophy that appear in adults are caused by tri- or tetranucleotide, e.g. (CTG)n and (CCTG)n, repeats where n may run into the thousands. The huge RNA transcripts that result interfere with the alternative splicing of other transcripts in the nucleus.
ALS is a neurodegenerative disorder leading to dementia and muscle weakness. (ALS is often called "Lou Gehrig's disease" after the baseball player who died from it.)
The most common mutation in ALS is an expansion of the number of repeats of the hexanucleotide GGGGCC in a gene on chromosome 9 from the normal two, or at least fewer than three dozen, to hundreds or even several thousand. Translation of both the sense and the antisense strands containing these repeats (and in all 3 reading frames; there is no ATG start codon) produces polymers with long strings of gly-ala, gly-pro, gly-arg (from the sense strand) as well as pro-ala, another pro-gly, and pro-arg from the antisense strand. These proteins, especially those containing arginine (arg) form aggregates that damage brain cells.
Duplications are a doubling of a section of the genome. During meiosis, crossing over between sister chromatids that are out of alignment can produce one chromatid with a duplicated gene and the other (not shown) with the two genes with deletions. In the case shown here, unequal crossing over created a second copy of a gene needed for the synthesis of the steroid hormone aldosterone.
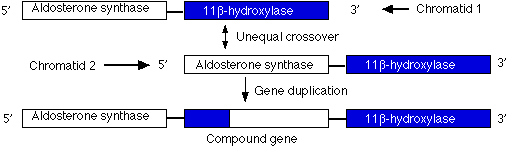
However, this new gene carries inappropriate promoters at its 5' end (acquired from the 11-beta hydroxylase gene) that cause it to be expressed more strongly than the normal gene. The mutant gene is dominant: all members of one family (through four generations) who inherited at least one chromosome carrying this duplication suffered from high blood pressure and were prone to early death from stroke.
Gene duplication has also been implicated in several human neurological disorders.
Gene duplication has occurred repeatedly during the evolution of eukaryotes. Genome analysis reveals many genes with similar sequences in a single organism. Presumably these paralogous genes have arisen by repeated duplication of an ancestral gene.
Such gene duplication can be beneficial.
Translocations are the transfer of a piece of one chromosome to a nonhomologous chromosome. Translocations are often reciprocal; that is, the two nonhomologues swap segments.
Translocations can alter the phenotype is several ways:
Mutations are rare events.
This is surprising. Humans inherit 3 x 109 base pairs of DNA from each parent. Just considering single-base substitutions, this means that each cell has 6 billion (6 x 109) different base pairs that can be the target of a substitution.
Single-base substitutions are most apt to occur when DNA is being copied; for eukaryotes that means during S phase of the cell cycle.
No process is 100% accurate. Even the most highly skilled typist will introduce errors when copying a manuscript. So it is with DNA replication. Like a conscientious typist, the cell does proofread the accuracy of its copy. But, even so, errors slip through.
It has been estimated that in humans and other mammals, uncorrected errors (= mutations) occur at the rate of about 1 in every 50 million (5 x 107) nucleotides added to the chain. (Not bad — I wish that I could type so accurately.) But with 6 x 109 base pairs in a human cell, that means that each new cell contains some 120 new mutations.
Should we be worried? The evidence is not clear.
Only 1.2% of our DNA encodes the exons of our proteome, and for a long time it was thought that much of the rest was "junk" DNA. Mutations in it would most likely be harmless. And even in coding regions, the existence of synonymous codons could result in the altered (mutated) gene still encoding the same amino acid in the protein.
But it now appears that as much as 80% of our DNA seems to participate in regulating which genes are expressed, and how strongly, in each of the multitude of differentiated cell types in our body as each responds to the signals (nutrients, hormones, etc.) it receives. [Link to a discussion.] So mutations in these regions might well have harmful, if subtle, effects.
As more vertebrate genomes are sequenced, it turns out that some of these stretches of DNA that do not encode proteins none-the-less have been remarkably conserved during vertebrate evolution. Some of these regions have accumulated even fewer mutations than protein-encoding genes have. This suggests that these sequences are extremely important to the welfare of the organism. However, other regions of the genome seem able to sustain point mutations with no detectible harm.
Recent advances have enabled the coding portions of the genome of single cells to be sequenced. Preliminary results indicate that each normal cell in an adult has accumulated ~20 somatic mutations, and that its collection of mutations differs from cell to cell. (Cancer cells accumulate many more mutations [often in the hundreds] — Link.)
How can we measure the frequency at which phenotype-altering mutations occur? In humans, it is not easy.But now these problems have been largely solved. The story is told in a report by D. R. Denver, et al. in the 5 August 2004 issue of Nature.
From these results I have pooled their data to calculate an approximate rate at which spontaneous mutations occur throughout the genome.
Mutation Rate = # of mutations observed [30] ÷ (# of experimental lines [198]) x (average # of generations [339]) x (average # of base pairs sequenced [~21,000])
yielding a rate of 2.1 x 10-8 mutations per base pair per generation.
The total C. elegans genome contains some 108 base pairs so this tells us that two new germline mutations occur somewhere in each of C. elegans's two haploid genomes in each generation.
A similar analysis for Drosophila (whose genome is about the same size as that of C. elegans) showed a similar mutation rate: ~10-8 mutations per base pair per generation. As for the green plant Arabidopsis thaliana, its spontaneous mutation rate is slightly lower: ~7 x 10-9 mutations per base pair per generation.
In the 30 April 2010 issue of Science, Roach, J. C., et al., reported that the rate for humans is in the same range: ~1.1 x 10-8 mutations per base pair in the haploid genome. With a diploid genome of 6 x 109 base pairs, that works out to some 70 new mutations in each child. They derived these numbers from comparing the complete genome sequence of two children and their parents.
In the 20 July 2012 issue of Cell, Wang, J., et al. reported the results of sequencing 8 individual sperm cells from a 40-year-old man. They found a mutation rate ranging from 2.0 x 10-8 to 3.8 x 10-8.
Should we be worried about such spontaneous mutation rates? Probably not too much. With our high proportion of noncoding DNA, many mutations will occur in regions that will have no effect on our phenotype. Evidence: out of a total of 251 mutations found in the 8 sperm cells, only 3 were missense mutations altering a gene product. However, even in noncoding DNA, point mutations may affect the expression of genes, so perhaps as many as 10% of the point mutations a child inherits may have harmful, if subtle, effects.
The significance of mutations is profoundly influenced by the distinction between germline and soma. Mutations that occur in a somatic cell, in the bone marrow or liver for example, may
Germline mutations, in contrast, will be found in every cell descended from the zygote to which that mutant gamete contributed. If an adult is successfully produced, every one of its cells will contain the mutation. Included among these will be the next generation of gametes, so if the owner is able to become a parent, that mutation will pass down to yet another generation.
| Link to a discussion of the distinctions between somatic cells and germline cells. |
| Ames Test for chemical mutagens |
| Measuring mutations in laboratory mice |
| Ionizing Radiation |
| Welcome&Next Search |
{kind=link}
{kind=link}